News & Announcements
Common Rule 2019
The revised Federal Policy for the Protection of Human Subjects (45 CFR 46 Subpart A) known as the Common Rule has been adopted by HHS and 15 Federal Agencies. The compliance date is January 21, 2019. More information can be found in the Federal Register.
In many cases, the Common Rule changes will not impact your ongoing research. In addition, the FDA has not yet agreed to the New Common Rule; therefore, changes outlined here will not be applicable to FDA regulated studies until we hear otherwise.
The Trinity Health Of New England IRB has made changes to relevant policies and procedures in accordance with the New Common Rule:
Revised Definitions: Research and Human Subjects
Informed Consent:
- New General Requirements
- New Elements
- Changes to the Waiver of Informed Consent
- Expansion of When Signed Informed Consent is not Required
Changes in IRB Review:
- Continuing Review - New Extended Approval Process (EAP)
- Limited Review
- Single IRB (sIRB)
Exempt Research
In many cases, the Common Rule changes will not impact your ongoing research. In addition, the FDA has not yet agreed to the New Common Rule; therefore, changes outlined here will not be applicable to FDA regulated studies until we hear otherwise.
If you have any questions, please contact the IRB office (860) 714-4068.
Revised Definitions
Research: The revised Common Rule adds a provision that identifies four types of activities as not being "research" as defined in the Rule:
- Certain scholarly and journalistic activities,
- Certain public health surveillance activities,
- Collection and analysis of information, specimens, or records, by or for a criminal justice agency for certain criminal justice or investigative purposes, and
- Certain authorized operational activities for national security purposes
45CFR46.102(l): Research means a systematic investigation, including research development, testing, and evaluation, designed to develop or contribute to generalizable knowledge.
Human Subject: The regulatory definition of human subject remains substantively the same but in the revised Common Rule "data" is replaced with "information or biospecimens" for clarity.
45CFR46.102(l): Human subject means a living individual about whom an investigator (whether professional or student) conducting research: (i) Obtains information or biospecimens through intervention or interaction with the individual, and, uses, studies, or analyzes the information or biospecimens; or (ii) Obtains, uses, studies, analyzes, or generates identifiable private information or identifiable biospecimens.
Informed Consent
New General Requirements:
Changes to the general requirements for informed consent to provide key information and promote autonomy by ensuring prospective subjects receive the information needed to make an informed decision.
Changes to the informed consent general requirements include:
- informed consent must give prospective subjects the information that a reasonable person would want to have in order to make an informed decision
- information must be presented in a way that facilitates an understanding of why one might, or might not, want to participate
- the informed consent form should not simply list isolated facts, but instead should help people process complicated information
- key information such as the study's purpose, risks, benefits, and alternatives, must be provided at the beginning of the consent form.
New Elements:
Four requirements have been added to the elements of informed consent.
The first is required for all studies, and is a statement about whether participants' information or biospecimens might (or will not) be stripped of identifiers and used for future research.
The three new elements of informed consent, to be added as applicable, are:
- information about possible commercial profit
- information about whether clinically relevant research results will be returned to the subjects
- information about whether research activities will or might include whole genome sequencing.
Changes to the Waiver of Informed Consent: When identifiable biospecimens or private information are involved, the IRB must determine that the research could not practicably be conducted without the use of the identifiable information. If the research could be done using non-identifiable information, then that is what should be done.
Expansion of When Signed Informed Consent is not Required: The IRB may waive the requirement for a signed informed consent form when the subjects are members of a distinct cultural group or community in which signing forms is not the norm, and the research involves no more than minimal risk, and there is an alternative method for documenting that consent was obtained.
Changes in IRB Review
Continuing Review - New Extended Approval Process (EAP) - Some minimal risk studies will no longer require annual review. The IRB has begun identifying applicable studies.
- IRB Approval letters and consent forms (when applicable) will document that the study qualifies for extended approval under the Common Rule, and therefore will not expire in the future.
- Extended approval will still require modifications be submitted for IRB approval prior to implementing changes; and events that meet prompt reporting criteria must be reported to the IRB in 5-10 days.
- Initially, extended approval will not be considered for studies that involve:
º Federal Funding
º VA
º FDA regulated components or FDA oversight
º Prisoners
º Stem Cells or SCRO protocols
º Other IRB issues
When the new Common Rule becomes effective on January 21, 2019, the extended approval process will be expanded to include minimal risk federally funded studies as well as minimal risk studies involving Medical Experimentation.
Limited IRB Review - Limited IRB review is a process that is required only for certain exemptions. In limited IRB review, the IRB must determine that certain conditions, primarily that adequate provisions are in place to protect the privacy of subjects and maintain confidentiality of the data, are met.
Single IRB (sIRB) - Most federally funded collaborative research projects based in the US will be required to use a sIRB effective January 2020. (As of January 25, 2018, NIH policy has required the use of a sIRB in accordance with NIH Policy.
In many cases, the Common Rule changes will not impact your ongoing research. In addition, the FDA has not yet agreed to the New Common Rule; therefore, changes outlined here will not be applicable to FDA regulated studies until we hear otherwise.
If you have any questions, please contact the IRB office – (860) 714-4068
Exemptions - Broadened and new Exempt categories:
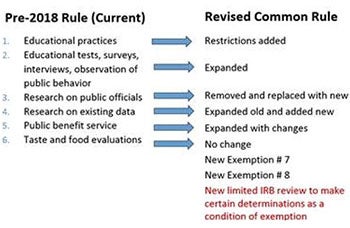
Category 1 (Educational Practices): research in established or commonly accepted educational settings that involves certain normal educational practices, such as research on instructional techniques already in use or classroom management. A new restriction to the applicability of Exemption 1 requires that the research must also not be likely to adversely impact the student's opportunity to learn required educational content or the assessment of educators who provide the instruction.
Category 2 (Educational tests, surveys, interviews, observation of public behavior): there are three primary changes:
- Addition that this "only includes interactions" involving educational tests, surveys, interviews, and observation of public behavior. Exemption 2 is not applicable to research involving interventions.
- Addition to second criterion requiring that the disclosure of the subjects' responses outside the research would not reasonably be damaging to the subjects' "educational advancement."
- Allows for the use of the limited IRB review where identifiable information (even if sensitive is recorded).
Category 3 (Benign Behavioral Interventions): research involving benign behavioral interventions with adults who prospectively agree to the research, when the information collected is limited to verbal or written responses, including data entry or audiovisual recordings. A benign behavioral intervention must be brief in duration (although data collection may take longer). Also, the intervention must be harmless, painless, and not physically invasive. Further, the intervention must not be likely to have a significant adverse lasting impact on subjects. The investigator must have no reason to believe that the intervention will be offensive or embarrassing to subjects, and should take into consideration the subjects' population, the context of the research, the topic, and other characteristics of the study.
Category 4 (Research on existing data): secondary research use of identifiable private information or identifiable biospecimens. One change in the revised Common Rule is that the private information and biospecimens no longer have to be in existence prior to the start of the research but must meet one of the applicability provisions.
Category 5 (Public benefit service): expanded to include research that is also supported by a federal department or agency (for example, through a grant of funding). There is also a new requirement for the federal entity conducting or sponsoring the research to publish a publicly available list of the projects that are covered by this exemption before the research begins.
Category 6 (Taste and Food Evaluations): No changes to this category that allows for the review or research involving taste and food quality evaluation and consumer acceptance studies.
Category 7 (Storage and maintenance for secondary research collected under broad consent): covers the storage or maintenance of identifiable private information or identifiable biospecimens for secondary research. Secondary research refers to research with materials originally obtained for nonresearch purposes or for research other than the current research proposal. The exemption can only be used when there is broad consent from the subjects for the storage, maintenance, and secondary research use of their identifiable materials.
Category 8 (Secondary research for which broad consent is required): covers the secondary research use of identifiable private information or identifiable biospecimens originally obtained for nonresearch purposes or for research other than the current proposal. There are four requirements that must be satisfied:
- broad consent must be obtained from the subjects for the secondary research use of their identifiable materials,
- documentation or waiver of documentation of informed consent must be obtained,
An IRB must conduct a limited review to make certain determinations relating to privacy and confidentiality protections and broad consent, and investigators cannot include the return of individual research results to subjects in the study plan. Note that this requirement does not limit an investigator's ability to abide by any other legal requirement to return individual research results.